© 2026 太和县人民医院 地址:安徽省太和县健康路21号
24小时投诉监督服务电话:0558-96511
一、安全性信息报告工作指引
递交要求
1.本中心注册类药物临床试验安全性事件
(1)研究者在首次获知SAE后24小时内报告至医学伦理委员会及申办者,所有SAE需按照研究方案的规定随访,提交SAE随访报告和总结报告。妊娠事件按照SAE要求进行报告。
(2)申办者收到SAE报告后,应立即分析评估是否为可疑且非预期严重不良反应(SUSAR)。对于致死或者危及生命的SUSAR,申办者应在首次获知后尽快报告至伦理委员会,但不得超过7天,并在随后的8天内报告、完善随访信息(以申办者获知事件为0天计算)。非致死或者危及生命的SUSAR,申办者应在首次获知后15天内尽早报告至伦理委员会。
2.非本中心注册类药物临床试验安全性事件
申办者收到外院安全性信息后,以汇总报告的形式按季度提交(以申办者获知事件为0天计算)。
3.DSUR报告
申办者定期汇总DSUR并向我院伦理委员会报告,原则上报告周期不超过一年。
4.注册类医疗器械临床试验安全性事件
(1)本中心SAE:研究者在首次获知SAE后24小时内报告至医学伦理委员会及申办者,在获知SAE的新信息后,应当及时提供详尽、书面的随访报告或总结报告。
(2)非本中心SAE:申办者应当在获知死亡或者危及生命的临床试验医疗器械相关严重不良事件后7日内、获知非死亡或者非危及生命的试验医疗器相关严重不良事件和其他严重安全性风险信息后15日内,报告本中心伦理委员会。
5.研究者发起的临床研究安全性事件
研究者在首次获知SAE后24小时内报告至医学伦理委员会,在获知SAE的新信息后,应当及时提供详尽、书面的随访报告或总结报告。
6.其他重要的安全性信息
如有其他重要的安全性信息,应及时报告伦理委员会,如研究者手册更新、特别关注不良事件、数据安全监测报告等、以及其他可能影响研究参与者安全、可能影响临床试验实施、可能改变伦理委员会同意意见的安全性信息。
二、方案偏离报告工作指引
1.定期报告偏离方案类型
(1)为避免研究对受试者紧急危害的偏离方案。
(2)严重偏离方案:
研究纳入了不符合纳入标准或符合排除标准的受试者;
符合中止试验规定而未让受试者退出研究;
给予错误治疗或剂量;
给予方案禁止的合并用药;
可能对受试者的权益和安全、以及研究的科学性造成显著影响的情况。
(3)持续偏离方案(指同一研究人员的同一违规行为在被要求纠正后,再次发生)或者研究者不配合监查/稽查,或者对违规事件不予以纠正。
以上可能增加受试者风险或影响临床研究实施的偏离方案,要求在获知后1个月内向伦理委员会报告。
本中心安全性事件报告流程图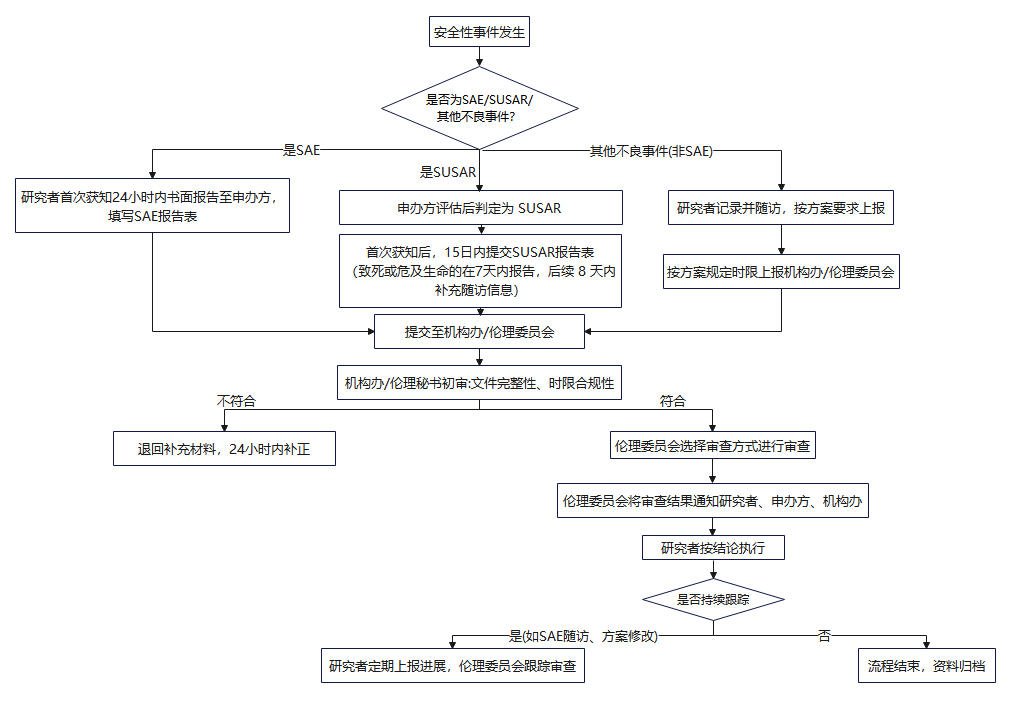
 皖公网安备 34122202000155号
皖公网安备 34122202000155号





